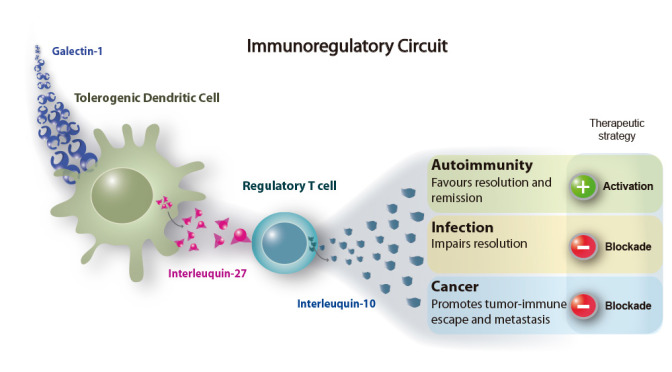
Despite their central function in orchestrating immunity, dendritic cells (DCs) can respond to inhibitory signals by becoming tolerogenic. Here we show that galectin-1, an endogenous glycan-binding protein, can endow DCs with tolerogenic potential. After exposure to galectin-1, DCs acquired an interleukin 27 (IL-27)-dependent regulatory function, promoted IL-10-mediated T cell tolerance and suppressed autoimmune inflammation of the central nervous system. Consistent with its regulatory function, galectin-1 had its highest expression on DCs exposed to tolerogenic stimuli and was most abundant from the peak through the resolution of autoimmune pathology. DCs lacking galectin-1 had greater immunogenic potential and an impaired ability to halt inflammatory disease.
Our findings identify a tolerogenic circuit linking galectin-1 signaling, IL-27-producing DCs and IL-10-secreting T cells, which has broad therapeutic implications in immunopathology.
Ilarregui et al. Nature Immunology, 2009 (see Publication section).
Despite the advances in the development of anti-angiogenic therapies, not every patient respond to the treatment, and those who initially were responders might become refractory. In this work, we found a link between hypoxia and neovascularization, which is dependent of galectin-1/glycan interactions in the tumor microenvironment, in Kaposi´s sarcoma. This interaction depends on NF-κB signaling but is independent of HIF-1α and HIF-2α. Targeted inhibition of galectin-1 prevents the formation of new blood vessels and tumor progression (Croci et al, Journal of Experimental Medicine, 2012).
Moreover, in recent studies we identified a new mechanism of resistance to anti-angiogenic therapies, through which binding of galectin-1 to complex N-glycans on VEFGR2, mimics VEGF signaling (Croci et al, Cell, 2014). The use of an anti-galectin-1 monoclonal antibody avoids tumor refractoriness, normalizes the tumor aberrant vasculature and potentiates the anti tumor immune response. The combination of these effects contributes to tumor growth inhibition.
Croci et al. Journal of Experimental Medicine, 2012.
Croci et al. Cell, 2014. (See Publications section)
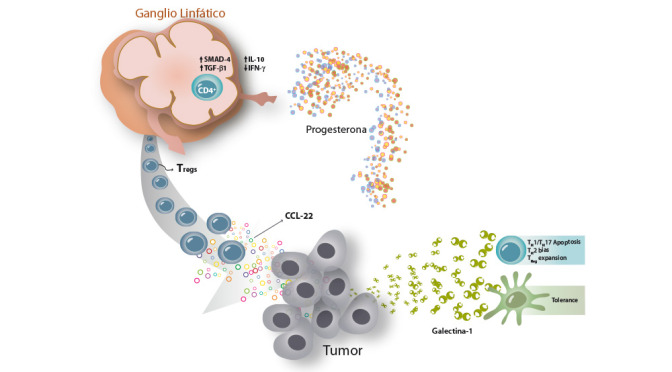
In this project we focus in the study of the tumor microenvironment in breast cancer; especially we investigate how interactions between cancer and immune cells determine the outcome of the metastasic process. In this regard, we recently demonstrated that galectin-1 is a key factor that promotes lung metastasis.Thus, blocking galectin-1-glycan interactions in the tumor microenvironment could be considered an effective strategy for treating metastatic breast cancer. Moreover, we are studying the capacity of steroid hormones to regulate the tumor microenvironment and their implications in tumor-immune escape.
We are also interested in elucidating how immune cell populations with tolerogenic activity can regulate the metastatic process. The information obtained in our work will set the fundamental bases for development of a third generation anticancer drugs aimed at blocking tumor immune escape, resistance to therapies and metastasis in breast cancer.
D’alotto Moreno et al, Cancer Research, 2013 (See Publications section).
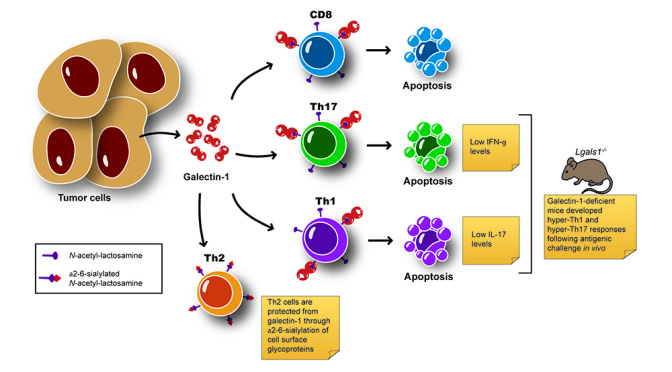
Regulated glycosylation can control T cell processes, including activation, differentiation and homing, by creating or masking ligands for endogenous lectins. Here we show that TH1, TH2 and TH-17-promoting stimuli can differentially regulate the glycosylation pattern of T helper cells and modulate their susceptibility to Gal-1, a glycan-binding protein with anti-inflammatory activity.
While TH1- and TH-17-differentiated cells express the repertoire of cell surface glycans that are critical for galectin-1-induced cell death, TH2 cells are protected from Gal-1 through differential sialylation of cell surface glycoproteins.
Consistently, Gal-1-deficient mice develop increased TH1 and TH-17 responses and show greatly enhanced susceptibility to autoimmune diseases. Our findings identify a molecular link between differential glycosylation of T helper cells, susceptibility to cell death and termination of the inflammatory response.
Toscana et al. Nature Immunology, 2007 (See Publications section).





